一患儿为何腹胀难治?骨髓象发现戈谢细胞
【前言】
戈谢细胞(Gaucher′s cell),又称高雪氏细胞,法国皮肤科医生Gaucher P1882年首先报道,是因溶酶体内的酸性β-葡萄糖苷酶(acid β-glucosidase),又称 葡萄糖脑苷脂酶(glucocerebrosidase,GC)缺陷导致戈谢病(Gaucher′s disease,GD,又称高雪氏病,高雪病),使葡萄糖脑苷脂贮积在各器官的单核巨噬细胞系统中,形成戈谢细胞。
【案例经过】
患儿,女,8岁,3个月前无明显诱因出现腹痛,以脐周疼痛为主,可自行缓解家属末予处理。1周前开始出现腹胀,无恶心、呕吐,无腹痛,无反酸、烧心,无厌食。今为进一步治疗,来我院急诊就诊拟“腹胀”收入儿科。发病以来,患儿精神、胃口一般,大小便正常。神志清,精神可,全身皮肤无皮疹、黄染,未见出血点及瘀斑、瘀点,无肝掌、蜘蛛痣,全身浅表淋巴结未扪及肿大。入院后完善辅助检查:
血常规检查(见图1)
白细胞(WBC),5.2x109/L,中性粒细胞百分数(NE%),40.8%,淋巴细胞百分数(LY%),51.3%,单核细胞百分数(MO%),4.0%,红细胞(RBC),4.48x1012/L,血红蛋白浓度(HGB),120g/L,血小板(PLT),93x109/L。
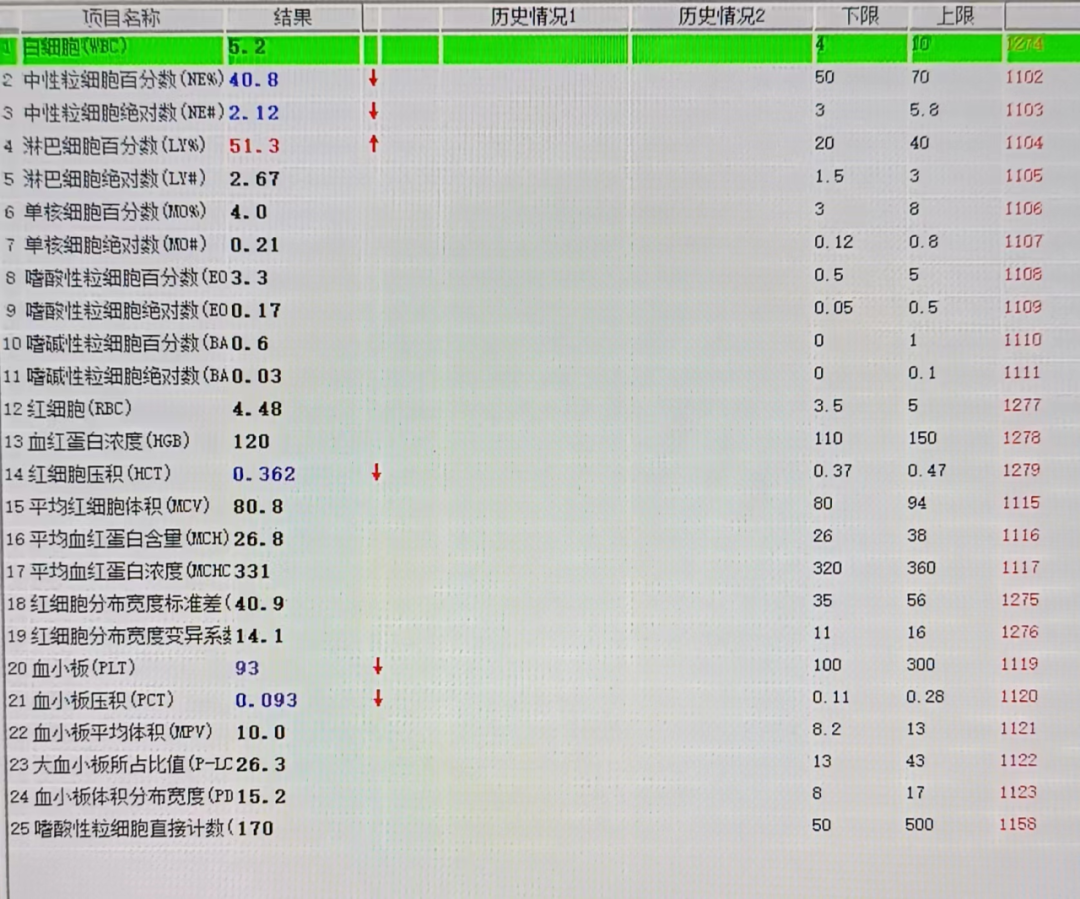
(图1)
凝血功能五项测定(见图2)
结果显示活化部分凝血活酶时间,46s。

(图2)
生化血酯检查(见图3)
载脂蛋白AI(APAI),0.70g/L,总胆固醇(CHOL),2.66mmol/L,甘油三酯(TG),2.38mmol/L。

(图3)
肝功8+肾功4+心肌酶5+CRP(见图4)
天冬氨酸氨基转移酶(AST)41.0U/L,乳酸脱氢酶(LDH)357.0U/L,总蛋白(TP)87.2g/L,球蛋白(GLB)43.2g/L。

(图4)
尿常规(见图5)
尿液白细胞酶(WBC),1+Cells/uL。

(图5)
免疫功能五项(见图6)
补体C3含量测定(C3),0.75g/L,补体c合量测定(C4),0.04g/L,IgM合量测定(IgM,5.40g/L,IgG含量测定(IgG),28.08g/L;血清铁蛋白测定:铁蛋白266.00ug/L。

(图6)
自身抗体定量未见异常(图7)
肝炎病毒全套未见异常(图8)

(图7)、(图8)
骨髓穿刺检查(见图9)
骨髓诊断意见:骨髓增生明显活跃,粒红两系均增生,可见疑似戈谢细胞。
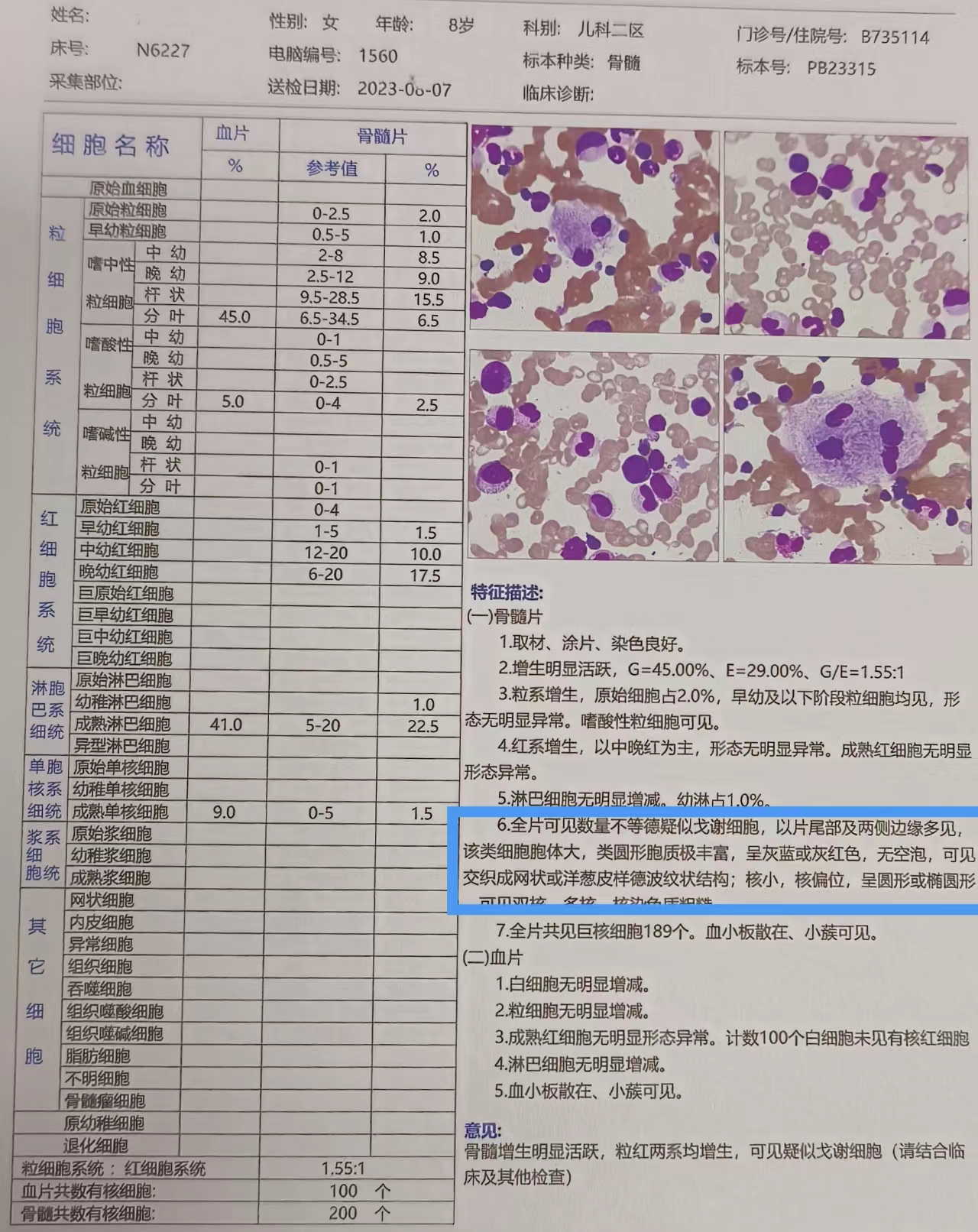
(图9)
【病例分析】
患儿因“反复腹痛3个月,腹胀1周”入院,查体肝脾明显肿大;实验检查白细胞升高,血小板减少,淋巴细胞比例大于中性粒细胞比例,肝功能、血脂未见明显异常。患儿身高112cm,3SD体重20kg,-2SD~=3SD,头围48cm,均较正常同龄儿童小。骨髓形态学检查:骨髓增生明显活跃,粒红两系均增生,骨髓片可见数量不等的疑似戈谢细胞,以片尾部及两侧边缘多见,此类细胞胞体大,直径20-80 um,细胞呈卵圆形或多边不规则形;胞核圆或椭圆形,较小,1-3 个或更多,多位于胞体的边缘,染色质粗糙,胞质丰富,淡蓝色,无空泡,胞质中含有许多与细胞长轴平行的粗暗条纹样结构,交织成网,如“洋葱皮”或“蜘蛛网”样(见下图)。患儿住院后对症治疗,效果不佳,不排除遗传性脂质代谢障碍性疾病。

【知识拓展】
戈谢病需与骨髓中见到戈谢样(吞噬)细胞的其他疾病鉴别,如慢性粒细胞白血病、免疫性血小板减少症、慢性淋巴细胞白血病、恶性淋巴瘤、多发髓瘤、骨髓增生异常综合征等,这些疾病有可能见到戈谢样吞噬细胞(见下图)。
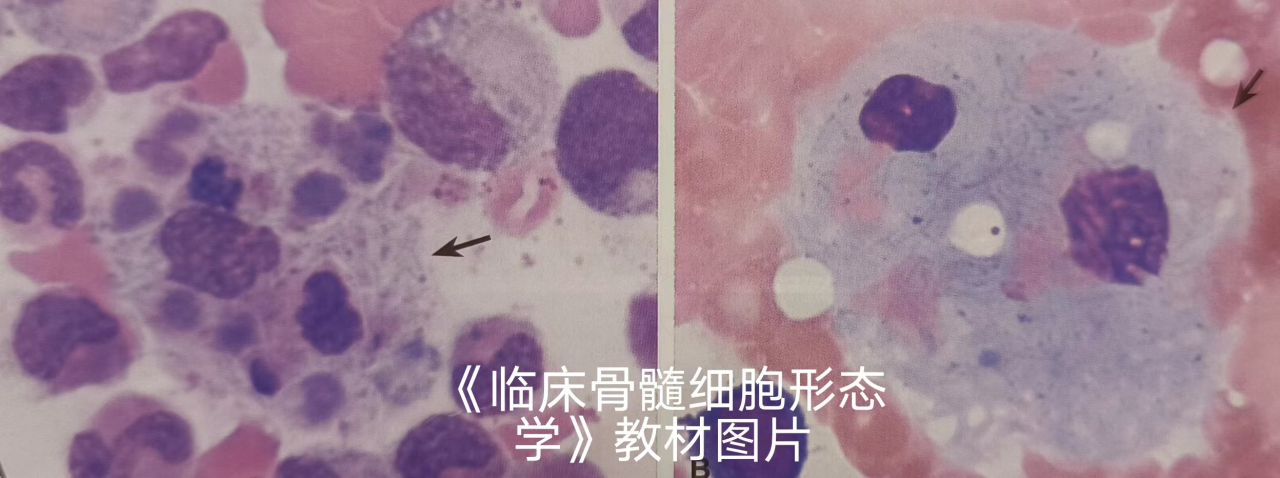
其他疾病骨髓中的戈谢样吞噬细胞与戈谢细胞在形态上较难区别,必须结合临床有否肝脾肿大、戈谢样细胞量、是否有基础疾病、“戈谢样细胞”的吞噬物情况等辅助鉴别[1]。
值得一提的是,尼曼-匹克病也为常染色体隐性遗传溶酶体贮积病,与戈谢病一样,临床皆可出现肝脾大、血细胞减少、骨质破坏等[2]。戈谢细胞与尼曼-匹克细胞的鉴别如下表:

两者相关酶学检测及基因突变检查可确诊并可鉴别。GBA活性检测明显降低或缺乏是戈谢病诊断的金标准,NPD为神经鞘磷脂酶缺失或活性下降,引起神经鞘磷脂沉积。
【总结】
在戈谢病中,贫血、血小板或白细胞减少,戈谢细胞可在骨髓中发现,胞体大或巨大,核小,有时可见核畸形、双核及多核,染色质致密;胞浆量丰富,呈灰蓝色“洋葱皮”样排列。戈谢细胞酸性磷酸酶活性升高,必要时可进行酸性磷酸酶染色。不过戈谢细胞并不是戈谢病特有的,亦可见于一些血液系统疾病及感染性疾病,此时称为“类戈谢细胞”,在慢性粒细胞白血病、地中海贫血、骨髓增生异常综合征、多发性骨髓瘤中均可能出现这种“类戈谢细胞”。
因此,当骨髓中存在戈谢细胞时,虽应高度怀疑GD,但需进一步行GBA活性测定以确诊,所以说,GBA活性检测明显降低或缺乏是戈谢病诊断的金标准[3]。
由于戈谢病属于罕见病,且具有确诊能力的实验室数量不多及治疗费用较高[4],所以外周血液及骨髓涂片检查找到一定数量的疑似戈谢细胞为戈谢病的诊断提供新思路和参考价值 。戈谢病在各年龄段均可受累,常于幼年期发病,病情呈进行性加重,如不尽早诊断以启动有效治疗,该疾病可导致不可逆的器官系统损害乃至残疾,甚至死亡[5]。戈谢病的临床治疗有酶替代治疗,底物减少治疗,造血干细胞移植,非特异性治疗,基因治疗等,随着对戈谢病研究的深入,国内外戈谢病的临床治疗方面涌现了许多新的研究成果,使戈谢病由不治之症成为了可以治疗的疾病。所以已建议该患者及时到上级医院完善确诊实验,接受最佳治疗。
参考文献:
[1] 王霄霞,夏薇,龚道元.临床骨髓细胞检验形态学[M].第一版.北京:人民卫生出版社.2019.217-218页
[2] 董欣,张贝克,张莹楠,等. 成年人戈谢病伴骨髓戈谢细胞及尼曼匹克细胞样泡沫细胞一例并文献复习[J]. 白血病·淋巴瘤,2021,30(4):239-241.
[3] 汪海青,尼曼-匹克病、戈谢病的实验诊断分析[J].中国实验诊断学,2012年1月第16卷第一期,155-156页.
[4] 苑晓舟.孟岩.断晋燕.王成彬。血常规和血脂指标诊断戈谢病的价值.疑难病杂志[J].2016年6月第15卷第6期,614-620页.
[5] 中华医学会血液学分会红细胞疾病(贫血)学组. 中国成人戈谢病诊治专家共识(2020)[J].中华医学杂志 2020年100卷24期, 1841-1849页.
作者:路引枝(三江侗族自治县人民医院检验科)
编辑:种一棵树

相关文章
热门文章

热门问题

